
生命科学をやっている人なら一度は出したことはある「シークエンス」。
構築したプラスミドの配列が正しいかどうか?ゲノム編集した部分がきちんとゲノム編集されているかどうか?など目では見えない塩基配列を解析する時には「シークエンスに出す」は常識ですよね。
その「シークエンス」、どうやって「読まれているか」ご存知ですか?きちんと説明できますか?
実はその「シークエンス」は大学の授業で必ず習う、「サンガー法」が用いられているのです。
次世代シークエンサーとかでバンバン配列が読める時代に、教科書に載るレベルのちょっと古典的な方法で解析されているって不思議ですよね。
そこで今回は、シークエンスに出す人は知っておきたい「サンガー法」についてまとめます。
「サンガー法」を知る前に知っておきたい知識
サンガー法を知る前に、「DNA合成反応」がどのように進むのかを知っておきましょう。
DNAは4種類の基質(dNTP[デオキシヌクレオチド三リン酸]:dATP, dGTP, dCTP, dTTP)が脱水縮合してつながっています。

DNA合成反応は一本鎖DNAを鋳型にしてDNAポリメラーゼが5'から3'の方向に鋳型DNAに相補的な配列を合成していきます。その際、短鎖RNAまたはDNA(プライマー)を起点として伸長反応が進みます。

これを試験管内(in vitro)でやっているのが、PCRです。
このDNA合成反応の過程を使って、DNAの塩基配列を「サンガー法」で決定していきます。
「サンガー法」の名前の由来になっているフレデリック・サンガーはロバート・ギルバートと共に、DNAシークエンス技術の開発の功績が認められて1980年(40年前)にノーベル賞を受賞しています。
「サンガー法」の鍵はddNTP
「サンガー法」の鍵となるのが、伸長反応を停止させるddNTP(2', 3'-ジデオキシヌクレオチド)です。
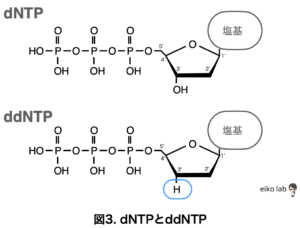
ddNTPは3'部位の-OH基が-H基に置換されています。そのため、脱水縮合反応を起こせず、次のdNTPが結合できないので伸長反応が停止してしまうと言う原理です。
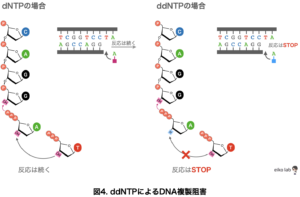
DNA合成反応液の中にddNTPのいずれか(ddATP, ddGTP, ddCTP, ddTTP)を入れておくと、ddNTPが取り込まれた時点で反応がストップします。この伸長反応停止がランダムに生じるため、様々な長さのDNA断片を得ることができます。
※例えば、ddATPを入れた反応系の場合、生じた断片の3'末端がAであることがわかります。
シークエンス用のサンプルの準備の仕方
昔は研究室でサンガー法を使ったシークエンスをやっていたようですが、最近は業者にお願いするところの方が多いと思います。
業者にシークエンスを出すときに準備するものは、
・配列を知りたいDNA(PCR産物・プラスミド)
・プライマー(Fwもしくは Rv)
を混ぜたものです。
(詳細な濃度に関しては、業者のHPなどを参照するようにしましょう)
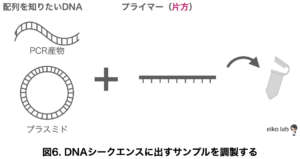
PCR産物をシークエンスに出す時には、PCRをかけた溶液をそのままシークエンスに出すことは望ましくないです。
その理由としては、
・PCR産物が均一(シングルバンド)ではない可能性がある
・残存dNTPがシークエンスに多少の影響を与える
から。
そのためにも、PCR産物をシークエンスに出す際はアガロースゲル電気泳動で目的のバンドを切り出してからサンプル調整をするようにしましょう。
アガロースゲル電気泳動・PCR産物の切り出し回収に関してはこちらの記事をご覧ください。

ちなみに、私の研究室の大学院生はこの「サンガー法」を不勉強なためにシークエンスを出す際にプライマーを二種類入れてしまうと言う大失態を犯しました...
プライマーを二種類入れると、両方向からシークエンスが読まれてしまうのでわけがわからなくなってしまいます。
サンガー法での配列決定の仕方
「サンガー法」はddNTPで反応をストップさせることで、様々な長さのDNA断片を作って配列を決定する方法でした。
では具体的にはどのように配列を決定するのでしょう?
昔は32P標識したdCTPなどを反応系に混ぜて、ポリアクリルアミドゲルで電気泳動した後、オートラジオグラフィーで観察していました。(古い研究室にはシークエンス用の薄くて大きなゲルを作るゲル板があったりします)
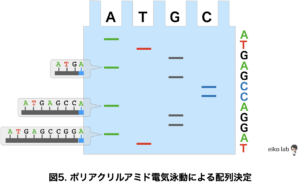
図5のようにddATPでストップさせたサンプル、ddTTPでストップさせたサンプル、ddGTPでストップさせたサンプル、ddCTPでストップさせたサンプルをそれぞれのレーンで流してDNA断片の長さの違いで配列を決定していました。
現在は、ddNTPそれぞれに違う色の蛍光をラベルしておいて、いずれか一つの蛍光で標識された様々な長さのDNA鎖ができます。それを一本鎖に解離して、キャピラリー電気泳動で長さの順に並べ、レーザーで検出を行うそうです(サンガーシークエンサー・DNA自動シークエンサー)。
DNAシークエンサーの仕組みについてはこちらの記事をご覧ください。
ApEを使ってシークエンス結果を見てみる
大抵は業者にお願いして、プライマー、鋳型DNAを混ぜた物を出すと.ab1ファイルや.fastaファイルなどでデータが返ってきますよね。
最後に、シークエンスの結果として送られてきた「.ab1」ファイルをApEを使ってみてみましょう。
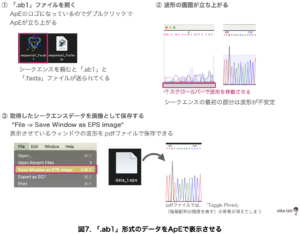
シークエンスのデータを画像データとして保存できるようになりました。
ApEのインストールに関してはこちらの記事をご覧ください。
ちなみに、シークエンスのデータを見てみると最初の20塩基と600塩基以降のシークエンスの精度が悪くなっています。
というのも、サンガー法を使ったDNAシークエンスではDNA鎖が長くなってくるとリード効率が落ちるからです。
シークエンスの精度はddNTPの取り込まれやすさに依存していて、ddNTPが取り込まれ辛い反応開始時とddNTPが少なくなってきた反応後半は検出効率が低くなるからではないかと思われます。
この「リード長」問題があるため、長い配列を確実に読みたい場合はプライマーを複数設計して複数のシークエンスデータを比較して(アライメント)配列を決定します。
もっと早く長いDNAの配列を読むためには「次世代シークエンサー」を使います。
「次世代シークエンサー」の原理についてはこちらの記事をご覧ください。
シークエンスを出す人は知らなきゃいけない?!「サンガー法」とは? まとめ
「サンガー法」についてまとめましょう。
- 「サンガー法」はddNTPを使うことでDNAの配列を決定する方法
- シークエンスに出す時にはプライマーは1種類のみを使うようにする
- 「.ab1」ファイルはApEを使って画像データにすることができる
- 「サンガー法」でシークエンスする時には「リード長」に注意する













